
Shi Pei Loo1, Kenneth Gilmour2, Hashnuhana Soma Chakrabarti3
1Foundation Doctor, Queen Elizabeth University Hospital, Glasgow, United Kingdom
2ST7 Ophthalmology Specialty Trainee, Department of Ophthalmology, Gartnavel General Hospital, Glasgow, United Kingdom
3Consultant Ophthalmologist, Department of Ophthalmology, New Victoria Hospital, Glasgow, United Kingdom
Learning Points
- VKH is uncommon in Caucasian and paediatric populations but it can occur.
- The lack of extraocular manifestations found in isolated ocular VKH can make the diagnosis more challenging.
- Prompt initiation of systemic corticosteroids and immunosuppression can result in excellent visual outcomes.
Background
Posterior uveitis is an uncommon cause of severe visual loss in the paediatric population, usually secondary to macular scarring or secondary glaucoma (1-3). Infectious aetiologies, such as toxoplasma and toxocara, are the most common and are usually associated with classical fundal examination findings (1, 3). Non-infectious causes are less common and can be a diagnostic challenge. We report an atypical presentation of an uncommon disease, particularly so in a paediatric population, with the potential for severe visual impairment if not diagnosed early and managed aggressively.
Case Presentation
A systemically well young teenage female with no ophthalmic or medical history presented with acute blurred vision in the right eye. Systemic enquiry was unremarkable, except for a history of viral illness causing cough and headache two months prior to presentation which the family also suffered from. This was assumed to be coronavirus disease (COVID-19), although no formal testing was available.
On examination, visual acuity (VA) was reduced to 0.700 Logmar in the right eye and subtly reduced to 0.040 Logmar in the left. The striking abnormality on fundal examination was extensive bilateral multifocal serous retinal detachments. Optical coherence tomography (OCT – Heidelberg spectralis) images in figure 1 demonstrate the right macular involvement, additionally the optic nerve head appeared swollen. There was no relative afferent pupillary defect or reduction in colour vision. There was no vitritis or anterior segment abnormality on presentation and the intraocular pressure was normal. However, upon review at one week, the patient had developed a bilateral anterior uveitis.
A uveitis screen was performed (table 1) and except for a positive HLA-DR4, the results were otherwise negative. OCT was performed at baseline (figure 1) and subsequent visits (figure 2) to monitor progression. OCT at presentation demonstrated bilateral multifocal high-sided serous retinal detachments (highest central subfield thickness (CST) 1054 microns). Additional findings included subretinal membranous structures, subretinal hyperreflective dots and retinal pigment epithelium (RPE) folds.
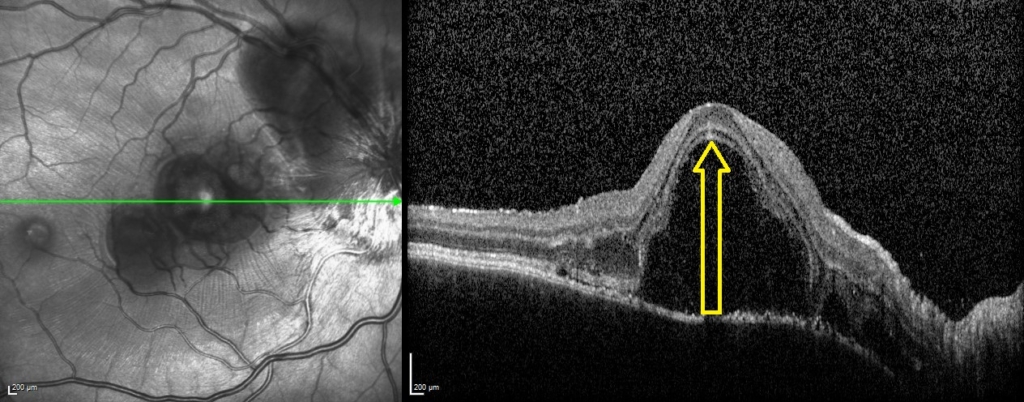
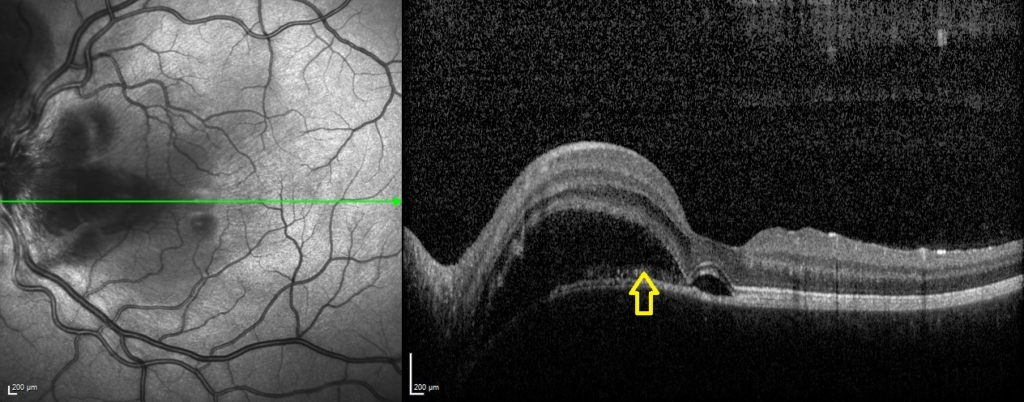
One week following presentation and initial treatment, early signs of improvement were evident with reduction in height of serous detachments (right CST 617 microns) although some macular involvement in the left eye had developed. Gradual resolution is evident on subsequent visits (figure 2), with a mild residual RPE disturbance. Fluorescein angiography and lumbar puncture are potentially valuable additional investigations which were considered but as OCT findings were characteristic, further invasive investigations in a child were deemed unnecessary.
Table 1: Investigations in paediatric uveitis screen and results
| Category | Investigation | Result |
|---|---|---|
| Haematology / Biochemistry | Full blood count | Normal |
| Erythrocyte sedimentation rate | Normal | |
| C reactive protein | Normal | |
| Renal Function | Normal | |
| Liver Function | Normal | |
| Angiotensin converting enzyme (ACE) levels | Normal | |
| Chitotriosidase levels | Normal | |
| Virology | Treponemal-syphilis antibody | Negative |
| Tuberculosis (TB) Quantiferon Gold Test | Negative | |
| Toxoplasma IgG | Negative | |
| Varicella zoster IgG | Detected (evidence of past infection/vaccination) | |
| Immunology | Antinuclear antibody (ANA) screen | Negative |
| Myeloperoxidase (MPO) antibodies | Negative | |
| Proteinase 3 (PR3) antibodies | Negative | |
| HLA Typing | HLA-B27 | Negative |
| HLA-B51 | Negative | |
| HLA-DRB1*04 | Negative | |
| HLA-DR4 | Positive | |
| Imaging | Chest Xray | Normal |
| MRI Head and Orbits | Normal |
An acute reduction in vision in an otherwise healthy patient with posterior segment findings was treated as posterior uveitis of unknown cause. The lack of a defining fundal lesion, bilateral presentation and widespread exudative detachments excluded more common infectious causes such as toxoplasmosis. Due to the presence of optic nerve head swelling, neuroretinitis was considered however there was no macular exudate formation and a bilateral presentation would be atypical. Other causes of panuveitis including tuberculosis, syphilis and sarcoidosis would be unusual in this age group and geographic location.
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE), posterior scleritis, central serous chorioretinopathy (CSCR), sympathetic ophthalmia and Vogt-Koyanagi-Harada disease (VKH) should all be considered. APMPPE may present similarly but was less likely based on the development of bilateral anterior uveitis and the absence of characteristic yellow or grey-white placoid lesions.
Pain is a hallmark feature of scleritis and was absent in this case, as was any history of prior ocular trauma or surgery, which is necessary for a diagnosis of sympathetic ophthalmia. Both CSCR and VKH can present with bilateral serous retinal detachments, and retrospective studies have shown high rates of misdiagnosis, with one large review finding 90/410 VKH cases initially diagnosed as CSCR (4). Although such marked bilateral findings would be atypical for CSCR, the distinction is important as corticosteroids potentially cause and worsen CSCR, but are recommended as first line management in VKH.
Discussion with adult uveitis specialists and examination of OCT showed correlation with highly specific OCT findings documented in the academic literature, and based on the Revised Diagnostic Criteria, isolated ocular VKH was diagnosed (5, 6).
Initial treatment of three consecutive daily doses of 1 gram intravenous (IV) methylprednisolone was commenced followed by a switch to oral prednisolone 1mg/kg and commencement of oral tacrolimus 50mcg/kg. Liaison with the paediatric rheumatology team provided access to paediatric nurse specialists with expertise in the use of immunosuppressive medications, blood monitoring, administration of medication, family and patient support as well as thorough investigation of potential systemic associations, including baseline audiological assessment.
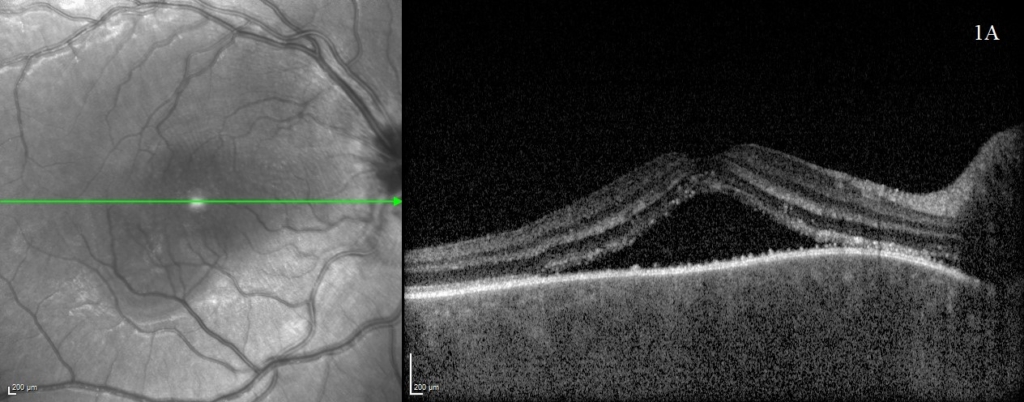
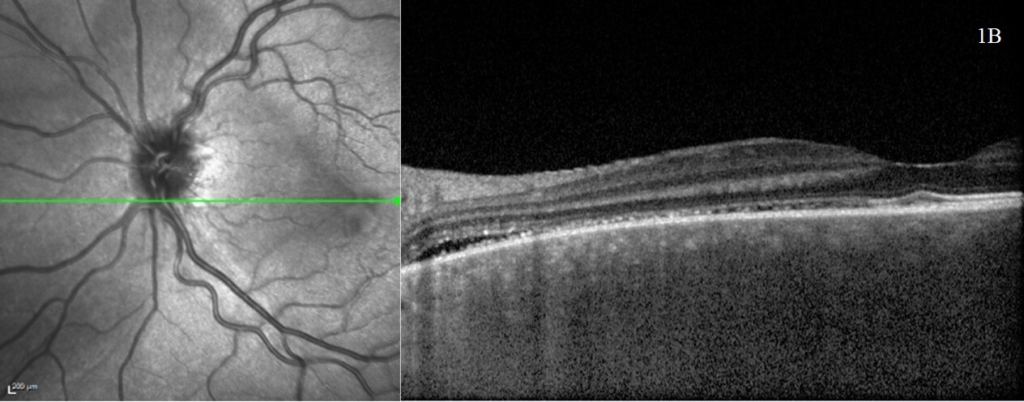
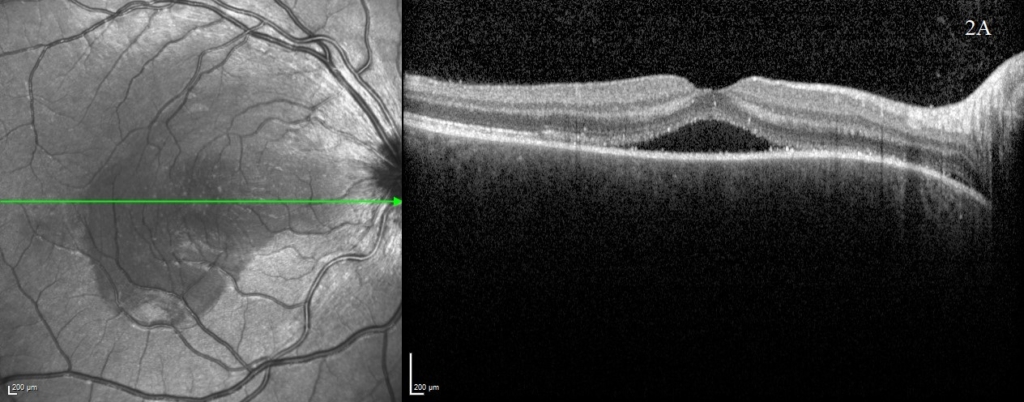
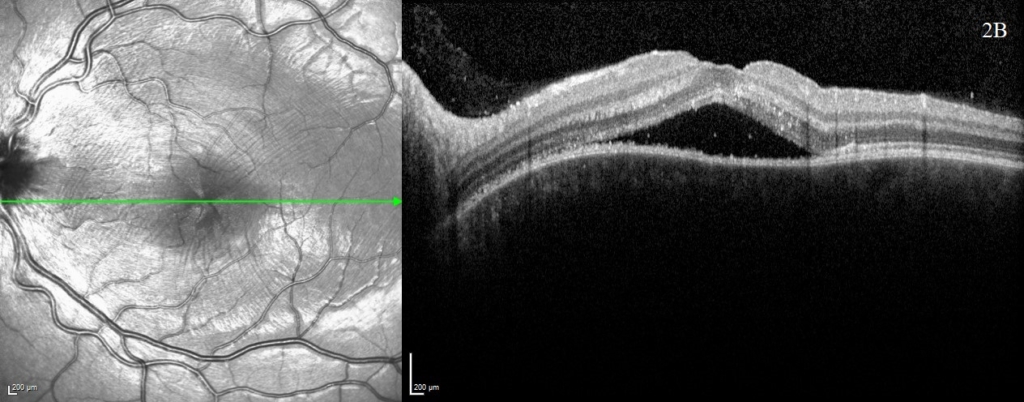
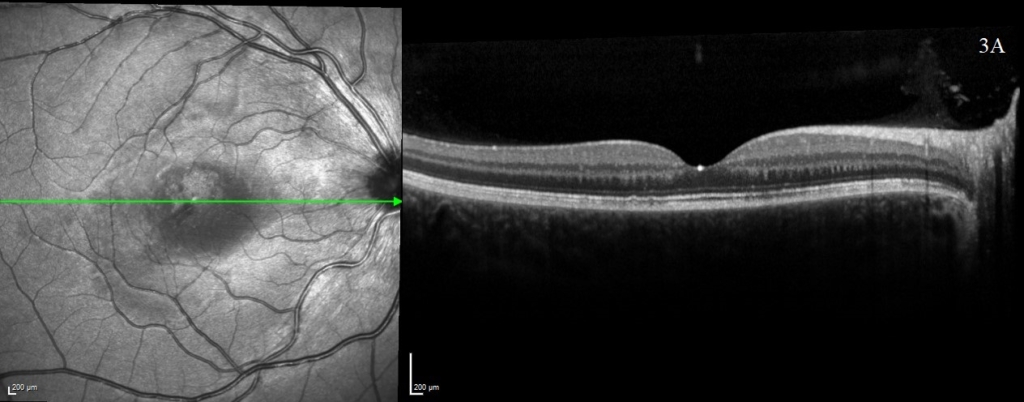
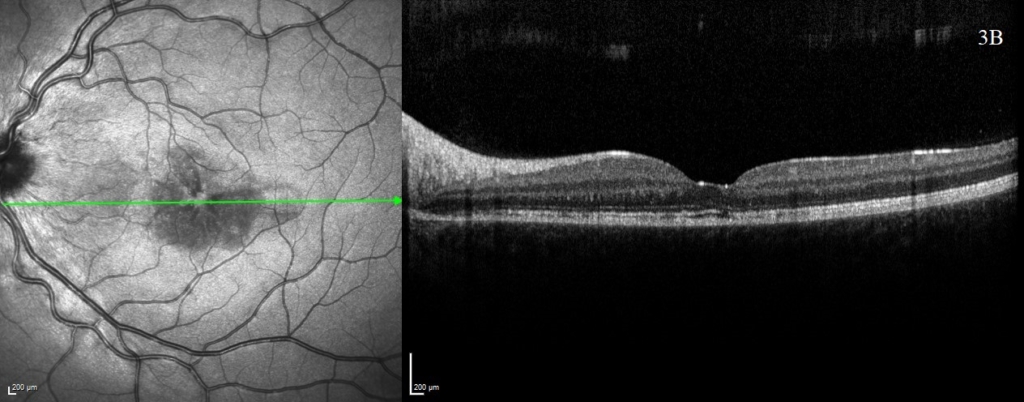
A rapid improvement in vision and OCT findings was observed following IV methylprednisolone (figure 2) and oral prednisolone sustained this improvement over subsequent weeks. Gradual resolution of serous detachments on OCT matched improvement in visual acuity with 0.000 Logmar vision observed in both eyes at week 4. Frequent reviews at a combined paediatric ophthalmology/rheumatology clinic allowed close monitoring. As per national recommendations, the patient was asked to shield during the COVID-19 pandemic.
After sustained control of symptoms over several months, oral prednisolone was tapered to 0.3mg/kg. Common side effects of corticosteroids were observed such as Cushingoid appearance and raised intraocular pressure managed with topical timolol. Thus far there has been no evidence of secondary glaucoma or cataract formation. No systemic manifestations of VKH have been identified in this patient after six months of follow up and interestingly, there has been no development of the classical secondary features associated with this condition. The long-term goal of treatment will aim to balance reduction of immunosuppressive medications whilst avoiding a recurrence.
Discussion
To our knowledge, there is currently no published literature on the management with tacrolimus of VKH in a Caucasian paediatric patient.
VKH is a bilateral non-necrotising diffuse granulomatous inflammation of the uveal tract resulting from a sporadically occurring T cell-mediated autoimmune response against melanocyte-rich tissues (7). The exact pathogenesis of VKH is unknown but patients often report a period of feeling unwell and associations with viral aetiologies have been reported, although no causative relationship exists (7).
In this case, the patient and family experienced a viral illness at the first peak of the UK COVID-19 pandemic. This was presumed to be COVID-19 although no formal testing was available and therefore COVID-19 cannot be shown to have been a prodromal illness in this case. However the authors thought it important to recognise that VKH occurred in an unusual patient demographic at the height of the COVID-19 pandemic and the effects of COVID-19 in the development of ophthalmic conditions remains in its infancy. Isolated case reports have reported VKH associations with COVID-19 and COVID-19 vaccination (8, 9).
VKH is associated with extraocular manifestations such as sensorineural hearing loss, tinnitus, vitiligo and neurological symptoms. Isolated ocular disease occurs in the absence of prior ocular trauma, systemic features or an alternative diagnosis. An uncommon condition, especially in the paediatric demographic, coupled with a lack of systemic symptoms made the diagnosis challenging. OCT was important in identifying specific features of VKH such as the presence of subretinal membranous structures, subretinal hyperreflective dots and RPE folds which have been demonstrated to have high sensitivity and specificity in diagnosing VKH (6).
VKH is very rare in northern European adult and paediatric ophthalmology settings and the exact prevalence is unknown. Retrospective studies consistently show VKH is more prevalent amongst pigmented ethnicities, particularly Asian populations, forming up to 16% of uveitis referrals in China and it most commonly presents between the second and fifth decades (7, 10-12). In the USA, a retrospective study of 65 adult cases found that only 3% were Caucasian (13). Epidemiological studies across northern European countries consistently find VKH represents less than 1% of uveitis referrals (7). Data regarding the prevalence of isolated ocular VKH as a proportion of VKH in children is sparse but may represent up to 31% of total VKH caseload (14).
There is no standardised treatment regimen for managing VKH in adult or paediatric populations. Paediatric VKH patients treated early and aggressively with corticosteroids tend to have better visual outcomes (15, 16). Poorer baseline visual acuity, longer duration of disease and increased frequency of recurrences are associated with poorer visual outcomes (17). Regarding immunosuppressive agents, there is a requirement for ongoing research, as there is a suggestion they may be effective as first line medications (7).
At present, management with systemic corticosteroids is considered first line management and prolonged courses of several months are recommended to prevent chronic recurrent forms of the condition from manifesting. The pathophysiology of VKH is considered to be a T-cell mediated response against pigment-containing tissues including the choroid, skin, ear and meninges (18). Both tacrolimus and ciclosporin selectively inhibit the T-cell mediated response by inhibiting calcineurin (14, 19). In this case tacrolimus was chosen with good effect due to the lower side effect profile. The authors hope that the excellent outcomes so far observed at six months follow-up can be sustained long term with the careful management and monitoring of immunosuppression.
Patient’s Perspective
When I was first diagnosed with VKH I felt relieved that the doctors knew why I was starting to lose my sight, but then I felt scared when I realised it was such a rare condition. When I was told I had to start on medication straight away I just kept hoping it would help, but felt a bit worried as I was told I would have to be on the highest dose of steroids along with other medication, which meant I had to shield (due to COVID 19).
I was told there could possibly be side effects but I would regard myself as quite lucky as I only really had restless legs, and struggled to sleep at night. The doctors also prepared me for the weight gain and how such a high dose of steroids could be dangerous for my bones, and recommended that I walk and exercise lots, which my family have helped me to do. The weight gain was the hard part, but I knew it was going to happen and it couldn’t be helped as I needed the steroids to help with my condition.
I quickly had to overcome my fear of needles as every time I visit the hospital I need to provide blood samples, which isn’t an easy task for me, as it always turns in to a drama for the nurses trying to get blood from me, as they can never find a vein. This isn’t a good combination, as I am also prone to fainting.
When the doctors told me my steroids could start to be reduced I was happy as this was happening much earlier than they had estimated, but I was very nervous in case it would start to flare things up again. I felt happy that the hospital were doing regular checks / scans on my eyes. I also had a hearing test, so I knew that if I started to develop problems with my ears they would be able to identify a change.
I have been lucky since reducing my steroids, although the pressure is still up in my eyes, and I get a little anxious if my eyes feel like they are changing, but reassured that I get regular scans and the doctors have told us to phone at any time.
References
1. Smith JA, Mackensen F, Sen HN, et al. Epidemiology and course of disease in childhood uveitis. Ophthalmology2009;116(8):1544-51,1551.e1 doi: 10.1016/j.ophtha.2009.05.002. Erratum for: Epidemiology and course of disease in childhood uveitis. Erratum in: Ophthalmology2011;118(8):1494.
2. de Boer J, Wulffraat N, Rothova A. Visual loss in uveitis of childhood. Br J Ophthalmol2003;87(7):879-84 doi: 10.1136/bjo.87.7.879.
3. Curragh DS, O’Neill M, McAvoy CE, et al. Pediatric Uveitis in a Well-Defined Population: Improved Outcomes with Immunosuppressive Therapy. Ocul Immunol Inflamm2018;26(6):978-985 doi: 10.1080/09273948.2017.1305420.
4. Yang P, Ren Y, Li B, et al. Clinical characteristics of Vogt-Koyanagi-Harada syndrome in Chinese patients. Ophthalmology2007;114(3):606-14 doi: 10.1016/j.ophtha.2006.07.040.
5. Salcedo HR, Feldman BH, O’Keefe GA, et al. Vogt-Koyanagi-Harada disease. American Academy of Ophthalmology; [Date of last update: 8 Mar 2021; Cited date: 27 Apr 2021]. Available from: https://eyewiki.aao.org/Vogt-Koyanagi-Harada_Disease.
6. Liu XY, Peng XY, Wang S, et al. Features of optical coherence tomography for the diagnosis of Vogt-Koyanagi-Harada disease. Retina2016;36(11):2116-2123 doi: 10.1097/IAE.0000000000001076.
7. Du L, Kijlstra A, Yang P. Vogt-Koyanagi-Harada disease: Novel insights into pathophysiology, diagnosis and treatment. Prog Retin Eye Res2016;52:84-111 doi: 10.1016/j.preteyeres.2016.02.002.
8. Yepez JB, Murati FA, Petitto M, et al. Vogt-Koyanagi-Harada Disease Following COVID-19 Infection. Case Rep Ophthalmol2021;12(3):804-808 doi: 10.1159/000518834.
9. Saraceno JJF, Souza GM, Dos Santos Finamor LP, et al. Vogt-Koyanagi-Harada Syndrome following COVID-19 and ChAdOx1 nCoV-19 (AZD1222) vaccine. Int J Retina Vitreous2021;7(1):49 doi: 10.1186/s40942-021-00319-3.
10. Goto H, Mochizuki M, Yamaki K, et al. Epidemiological Survey of Intraocular Inflammation in Japan. Jpn J Ophthalmol2007;51(1):41-4 doi: 10.1007/s10384-006-0383-4.
11. Ohguro N, Sonoda KH, Takeuchi M, et al. The 2009 prospective multi-center epidemiologic survey of uveitis in Japan. Jpn J Ophthalmol2012;56(5):432-5 doi: 10.1007/s10384-012-0158-z.
12. Yang P, Zhang Z, Zhou H, et al. Clinical patterns and characteristics of uveitis in a tertiary center for uveitis in China. Curr Eye Res2005;30(11):943-8doi: 10.1080/02713680500263606.
13. Moorthy RS, Inomata H, Rao NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol1995;39(4):265-92 doi: 10.1016/s0039-6257(05)80105-5.
14. Martin TD, Rathinam SR, Cunningham ET Jr. Prevalence, clinical characteristics, and causes of vision loss in children with Vogt-Koyanagi-Harada disease in South India. Retina2010;30(7):1113-21 doi: 10.1097/IAE.0b013e3181c96a87.
15. Yamanaka E, Ohguro N, Yamamoto S, et al. Evaluation of pulse corticosteroid therapy for vogt-koyanagi-harada disease assessed by optical coherence tomography. Am J Ophthalmol2002; 134(3):454-6 doi: 10.1016/s0002-9394(02)01575-1.
16. Yoshida A, Tominaga S, Kawashima H. Juvenile Vogt-Koyanagi-Harada Disease in Which Good Visual Prognosis Was Derived from Swift and Definitive Diagnosis. Case Rep Ophthalmol Med2016:7936729 doi: 10.1155/2016/7936729.
17. Read RW, Rechodouni A, Butani N, et al. Complications and prognostic factors in Vogt-Koyanagi-Harada disease. Am J Ophthalmol2001;131(5):599-606 doi: 10.1016/s0002-9394(01)00937-0.
18. Sugita S, Takase H, Taguchi C, et al. Ocular infiltrating CD4+ T cells from patients with Vogt-Koyanagi-Harada disease recognize human melanocyte antigens. Invest Ophthalmol Vis Sci2006;47(6):2547–54 doi: 10.1167/iovs.05-1547.
19. Cuchacovich M, Solanes F, Díaz G, et al. Comparison of the clinical efficacy of two different immunosuppressive regimens in patients with chronic vogt-koyanagi-harada disease. Ocul Immunol Inflamm2010;18(3):200-7 doi: 10.3109/09273941003587541.